First-Principles Simulations of Tip Enhanced Raman Scattering Reveal Active Role of Substrate on High-Resolution Images
Abstract
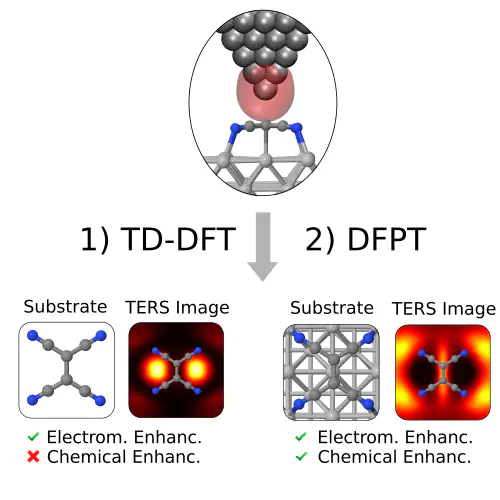
Tip-enhanced Raman scattering (TERS) has emerged as a powerful tool to obtain subnanometer spatial resolution fingerprints of atomic motion. Theoretical calculations that can simulate the Raman scattering process and provide an unambiguous interpretation of TERS images often rely on crude approximations of the local electric field. In this work, we present a novel and first principles-based method to compute TERS images by combining Time-Dependent Density Functional Theory (TD-DFT) and Density Functional Perturbation Theory (DFPT) to calculate Raman cross sections with realistic local fields. We present TERS results on the benzene and TCNE molecule, the latter of which is adsorbed at Ag(110). We demonstrate that chemical effects on chemisorbed molecules, often ignored in TERS simulations of medium and large systems sizes, dramatically change TERS images. This calls for the inclusion of chemical effects for predictive theory-experiment comparisons and understanding of molecular motion at the nanoscale.